Многоизмерни роли на кетонните тела
Кетонните тела се създават от черния дроб и се използват като източник на енергия, когато глюкозата не е лесно достъпна в човешкото тяло. Двете основни кетонни тела са ацетоацетат (AcAc) и 3-бета-хидроксибутират (3HB), докато ацетонът е третото и най-малко разпространено кетонно тяло. Кетоните винаги присъстват в кръвта и нивата им се повишават по време на гладуване и продължителни упражнения. Кетогенеза е биохимичният процес, чрез който организмите произвеждат кетонни тела чрез разграждането на мастни киселини и кетогенни аминокиселини.
Кетонните тела се генерират главно в митохондриите на чернодробните клетки. Кетогенезата възниква, когато има ниски нива на глюкоза в кръвта, особено след изчерпване на други клетъчни запаси от въглехидрати, като гликоген. Този механизъм може да възникне и при недостатъчни количества инсулин. Производството на кетонни тела в крайна сметка започва, за да направи достъпна енергия, която се съхранява в човешкото тяло като мастни киселини. Кетогенезата се случва в митохондриите, където се регулира независимо.
абстрактен
Метаболизмът на кетонните тела е централен възел във физиологичната хомеостаза. В този преглед ние обсъждаме как кетоните служат на отделни метаболитни роли за фина настройка, които оптимизират работата на органите и организма в различни остатъци от хранителни вещества и предпазват от възпаление и нараняване в множество органни системи. Традиционно разглеждани като метаболитни субстрати, включени само при ограничаване на въглехидратите, последните наблюдения подчертават важността на кетонните тела като жизненоважни метаболитни и сигнални медиатори, когато въглехидратите са в изобилие. Допълвайки репертоара от известни терапевтични възможности за заболявания на нервната система, се появиха перспективни роли на кетонните тела при рак, както и интригуващи защитни роли в сърцето и черния дроб, отваряйки терапевтични възможности при свързани със затлъстяването и сърдечно-съдови заболявания. Обсъждат се противоречия в кетонния метаболизъм и сигнализирането, за да се съгласуват класическата догма със съвременните наблюдения.
Въведение
Кетонните тела са жизненоважен алтернативен метаболитен източник на гориво за всички области на живота, еукария, бактерии и археи (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Метаболизмът на кетонните тела при хората е използван за подхранване на мозъка по време на епизодични периоди на недостиг на хранителни вещества. Кетонните тела са преплетени с важни метаболитни пътища при бозайниците като ?-окисление (FAO), цикъл на трикарбоксилната киселина (TCA), глюконеогенеза, de novo липогенеза (DNL) и биосинтеза на стероли. При бозайниците кетонните тела се произвеждат предимно в черния дроб от ацетил-КоА, извлечен от FAO, и се транспортират до екстрахепаталните тъкани за терминално окисление. Тази физиология осигурява алтернативно гориво, което се допълва от сравнително кратки периоди на гладуване, което увеличава наличността на мастни киселини и намалява наличността на въглехидрати (Cahill GF Jr, 2006; McGarry and Foster, 1980; Robinson and Williamson, 1980). Окислението на кетонните тела става значителен принос за цялостния енергиен метаболизъм на бозайниците в извънчернодробните тъкани в безброй физиологични състояния, включително гладуване, гладуване, неонаталния период, след тренировка, бременност и придържане към диети с ниско съдържание на въглехидрати. Концентрациите на циркулиращите общи кетонни тела при здрави възрастни хора обикновено показват циркадианни колебания между приблизително 100-250 µM, повишават се до ~1 mM след продължително упражнение или 24 часа гладуване и могат да се натрупват до 20 mM при патологични състояния като диабетна кетоацидоза ( Cahill GF Jr, 2006; Johnson et al., 1969b; Koeslag et al., 1980; Robinson and Williamson, 1980; Wildenhoff et al., 1974). Човешкият черен дроб произвежда до 300 g кетонни тела на ден (Balasse and Fery, 1989), които допринасят между 5% от общия разход на енергия в нахранени, гладни и гладни състояния (Balasse et al., 20; Cox et al. др., 1978).
Последните проучвания подчертават императивната роля на кетонните тела в метаболизма на клетките на бозайниците, хомеостазата и сигнализирането при голямо разнообразие от физиологични и патологични състояния. Освен че служат като енергийни горива за извънчернодробни тъкани като мозък, сърце или скелетни мускули, кетонните тела играят основна роля като сигнални медиатори, двигатели на протеинова пост-транслационна модификация (PTM) и модулатори на възпаление и оксидативен стрес. В този преглед ние предоставяме както класически, така и съвременни възгледи за плейотропните роли на кетонните тела и техния метаболизъм.
Преглед на метаболизма на кетонното тяло
Скоростта на чернодробната кетогенеза се управлява от организирана серия от физиологични и биохимични трансформации на мазнините. Първичните регулатори включват липолиза на мастни киселини от триацилглицероли, транспорт до и през плазмената мембрана на хепатоцитите, транспорт в митохондриите чрез карнитин палмитоилтрансфераза 1 (CPT1), β-окислителна спирала, активност на TCA цикъла и междинни концентрации, потенциал на редокс регулатор и хормонален регулаторен потенциал и хормонален регулаторен ефект. от тези процеси, предимно глюкагон и инсулин [прегледани в (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry and Foster , 1980; Williamson et al., 1969)]. Класически кетогенезата се разглежда като преливащ път, при който ацетил-КоА, получен от а-окисление, превишава активността на цитрат синтазата и/или наличността на оксалоацетат за кондензация за образуване на цитрат. Междинните съединения с три въглерода проявяват антикетогенна активност, вероятно поради способността им да разширяват оксалоацетатния пул за консумация на ацетил-CoA, но концентрацията на ацетил-CoA в черния дроб сама по себе си не определя кетогенната скорост (Foster, 1967; Rawat and Menahan, 1975; Williamson et al., 1969). Регулирането на кетогенезата чрез хормонални, транскрипционни и пост-транслационни събития заедно подкрепят идеята, че молекулярните механизми, които прецизират кетогенната скорост, остават ненапълно разбрани (вижте Регулация на HMGCS2 и SCOT/OXCT1).
Кетогенезата се извършва предимно в чернодробния митохондриален матрикс със скорост, пропорционална на общото окисление на мазнините. След транспортиране на ацилови вериги през митохондриалните мембрани и ?-окисление, митохондриалната изоформа на 3-хидроксиметилглутарил-CoA синтаза (HMGCS2) катализира съдбата, извършвайки кондензация на ацетоацетил-CoA (AcAc-CoA) и генериране на HMG-Ac-CoA (фиг. 1А). HMG-CoA лиаза (HMGCL) разцепва HMG-CoA, за да освободи ацетил-CoA и ацетоацетат (AcAc), а последният се редуцира до d-p-хидроксибутират (d-pOHB) от фосфатидилхолин-зависима митохондриална d-pOHB дехидрогеназа (D-pOHB дехидрогеназа). BDH1) в NAD+/NADH-свързана почти равновесна реакция (Bock and Fleischer, 1975; LEHNINGER et al., 1960). Равновесната константа на BDH1 благоприятства производството на d-?OHB, но съотношението на AcAc/d-?OHB кетонни тела е право пропорционално на съотношението NAD+/NADH на митохондриите и по този начин активността на BDH1 оксидоредуктазата модулира митохондриалния редокс потенциал (Krebs et al., 1969; Williamson et al., 1967). AcAc може също така спонтанно да се декарбоксилира до ацетон (Pedersen, 1929), източник на сладка миризма при хора, страдащи от кетоацидоза (т.е. общи серумни кетонни тела > ~7 mM; AcAc pKa 3.6, ?OHB pKa 4.7). Механизмите, чрез които кетонните тела се транспортират през вътрешната мембрана на митохондриите, не са известни, но AcAc/d-?OHB се освобождават от клетките чрез монокарбоксилатни транспортери (при бозайници, MCT 1 и 2, известни също като носители на разтворено вещество 16A членове 1 и 7) и транспортирани в циркулацията до екстрахепаталните тъкани за терминално окисление (Cotter et al., 2011; Halestrap and Wilson, 2012; Halestrap, 2012; Hugo et al., 2012). Концентрациите на циркулиращи кетонни тела са по-високи от тези в екстрахепаталните тъкани (Harrison and Long, 1940), което показва, че кетонните тела се транспортират надолу по градиент на концентрацията. Мутациите със загуба на функция в MCT1 са свързани със спонтанни пристъпи на кетоацидоза, което предполага критична роля във вноса на кетонни тела.

С изключение на потенциалното отклоняване на кетонните тела в неокислителни съдби (вижте Неокислителни метаболитни съдби на кетонните тела), хепатоцитите нямат способността да метаболизират кетонните тела, които произвеждат. Кетонните тела, синтезирани de novo от черния дроб, (i) се катаболизират в митохондриите на екстрахепаталните тъкани до ацетил-CoA, който е наличен в TCA цикъла за терминално окисление (фиг. 1A), (ii) се отклонява към пътищата на липогенезата или синтеза на стерол ( Фиг. 1В), или (iii) се екскретира с урината. Като алтернативно енергийно гориво, кетонните тела се окисляват жадно в сърцето, скелетните мускули и мозъка (Balasse and Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988 ). Екстрахепаталната митохондриална BDH1 катализира първата реакция на окисление на ?OHB, превръщайки я в обратно AcAc (LEHNINGER et al., 1960; Sandermann et al., 1986). Цитоплазмената d-?OHB-дехидрогеназа (BDH2) само с 20% идентичност на последователността с BDH1 има висок Km за кетонни тела и също така играе роля в хомеостазата на желязо (Davuluri et al., 2016; Guo et al., 2006) . В екстрахепаталния митохондриален матрикс, AcAc се активира до AcAc-CoA чрез обмен на CoA-част от сукцинил-CoA в реакция, катализирана от уникална CoA трансфераза на бозайници, сукцинил-CoA:3-оксокиселина-CoA трансфераза (SCOT, CoA трансфераза; кодиран от OXCT1), чрез реакция, близка до равновесие. Свободната енергия, освободена от хидролизата на AcAc-CoA, е по-голяма от тази на сукцинил-CoA, което благоприятства образуването на AcAc. По този начин окислителният поток на кетонните тела възниква поради масово действие: изобилното снабдяване с AcAc и бързата консумация на ацетил-CoA чрез цитрат синтаза благоприятства образуването на AcAc-CoA (+ сукцинат) от SCOT. По-специално, за разлика от глюкозата (хексокиназа) и мастните киселини (ацил-КоА синтетази), активирането на кетонните тела (SCOT) в окисляема форма не изисква инвестиция на АТФ. Обратима AcAc-CoA тиолазна реакция [катализирана от всяка от четирите митохондриални тиолази, кодирани от ACAA2 (кодиращ ензим, известен като T1 или CT), ACAT1 (кодиращ T2), HADHA или HADHB] дава две молекули ацетил-CoA които влизат в TCA цикъла (Hersh and Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). По време на кетозни състояния (т.е. общи серумни кетони > 500 M), кетонните тела допринасят значително за разхода на енергия – и се използват в тъканите бързо, докато настъпи поглъщане или насищане с окисляване (Balasse et al., 1978; Balasse and Fery, 1989 Edmond et al., 1987). Много малка част от извлечените от черния дроб кетонни тела могат лесно да бъдат измерени в урината, а степента на използване и реабсорбция от бъбреците са пропорционални на циркулиращата концентрация (Goldstein, 1987; Robinson and Williamson, 1980). По време на силно кетотични състояния (> 1 mM в плазмата), кетонурията служи като полуколичествен репортер на кетоза, въпреки че повечето клинични анализи на кетонни тела в урината откриват AcAc, но не и ?OHB (Klocker et al., 2013).
Кетогенни субстрати и тяхното влияние върху метаболизма на хепатоцитите
Кетогенните субстрати включват мастни киселини и аминокиселини (фиг. 1В). Катаболизмът на аминокиселините, особено на левцина, генерира около 4% от кетонните тела в постабсорбционно състояние (Thomas et al., 1982). Така ацетил-CoA субстратният пул за генериране на кетонни тела произлиза главно от мастни киселини, тъй като по време на състояния на намалено снабдяване с въглехидрати, пируватът влиза в чернодробния TCA цикъл предимно чрез анаплероза, т.е. АТФ-зависимо карбоксилиране до оксалоацетат (OAA) или до малат (MAL), а не окислително декарбоксилиране до ацетил-КоА (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). В черния дроб глюкозата и пируватът допринасят незначително за кетогенезата, дори когато декарбоксилирането на пируват до ацетил-КоА е максимално (Jeoung et al., 2012).
Ацетил-КоА включва няколко роли, които са неразделна част от чернодробния междинен метаболизъм извън генерирането на АТФ чрез терминално окисление (вижте също Интегрирането на метаболизма на кетоновите тела, пост-транслационната модификация и клетъчната физиология). Ацетил-КоА алостерично активира (i) пируват карбоксилаза (PC), като по този начин активира механизъм за метаболитен контрол, който увеличава анаплеротично навлизане на метаболити в TCA цикъла (Owen et al., 2002; Scrutton and Utter, 1967) и (ii) дехидрогеназа киназа, която фосфорилира и инхибира пируват дехидрогеназа (PDH) (Cooper et al., 1975), като по този начин допълнително засилва потока на пируват в TCA цикъла чрез анаплероза. Освен това, цитоплазменият ацетил-CoA, чийто пул се увеличава от механизми, които превръщат митохондриалния ацетил-CoA в транспортируеми метаболити, инхибира окисляването на мастни киселини: ацетил-CoA карбоксилазата (ACC) катализира превръщането на ацетил-CoA в малонил-CoA, липогенния субстрат и алостеричен инхибитор на митохондриален CPT1 [прегледан в (Kahn et al., 2005; McGarry and Foster, 1980)]. По този начин, митохондриалният пул ацетил-КоА едновременно регулира и се регулира от преливащия път на кетогенезата, който ръководи ключови аспекти на чернодробния междинен метаболизъм.
Неокислителни метаболитни съдби на кетонните тела
Доминиращата съдба на кетони, получени от черния дроб, е SCOT-зависимо екстрахепатално окисление. Въпреки това, AcAc може да бъде изнесен от митохондриите и използван в анаболни пътища чрез превръщане в AcAc-CoA чрез ATP-зависима реакция, катализирана от цитоплазмената ацетоацетил-CoA синтетаза (AACS, Фиг. 1B). Този път е активен по време на развитието на мозъка и в кърмещата млечна жлеза (Morris, 2005; Robinson and Williamson, 1978; Ohgami et al., 2003). AACS също е силно експресиран в мастната тъкан и активираните остеокласти (Aguilo et al., 2010; Yamasaki et al., 2016). Цитоплазмената AcAc-CoA може да бъде или насочена от цитозолен HMGCS1 към биосинтеза на стерол, или разцепена от една от двете цитоплазмени тиолази до ацетил-CoA (ACAA1 и ACAT2), карбоксилирана до малонил-CoA и да допринесе за синтеза на мастни киселини и др. al., 1984; Edmond, 1974; Endemann et al., 1982; Geelen et al., 1983; Webber и Edmond, 1977).
Докато физиологичното значение все още не е установено, кетоните могат да служат като анаболни субстрати дори в черния дроб. В изкуствен експериментален контекст, AcAc може да допринесе за до половината от новосинтезирания липид и до 75% от новия синтезиран холестерол (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Тъй като AcAc се получава от непълно окисление на чернодробните мазнини, способността на AcAc да допринесе за липогенезата in vivo би означавало безполезен чернодробен цикъл, при който кетони, получени от мазнини, могат да бъдат използвани за производството на липиди, понятие, чието физиологично значение изисква експериментално валидиране, но може да служи адаптивни или неадаптивни роли (Solinas et al., 2015). AcAc жадно осигурява холестерогенеза, с нисък AACS Km-AcAc (~50 µM), благоприятстващ активирането на AcAc дори в състояние на хранене (Bergstrom et al., 1984). Предполага се, че динамичната роля на метаболизма на цитоплазмения кетони в първичните миши ембрионални неврони и в адипоцитите, получени от 3T3-L1, тъй като нокдаунът на AACS нарушава диференциацията на всеки клетъчен тип (Hasegawa et al., 2012a; Hasegawa et al., ). Нокдаунът на AACS при мишки in vivo намалява серумния холестерол (Hasegawa et al., 2012c). SREBP-2012, главен транскрипционен регулатор на биосинтезата на холестерола и рецептор, активиран от пероксизомен пролифератор (PPAR)-? са AACS транскрипционни активатори и регулират транскрипцията му по време на развитието на неврит и в черния дроб (Aguilo et al., 2; Hasegawa et al., 2010c). Взети заедно, метаболизмът на цитоплазменото кетонно тяло може да бъде важен при избрани състояния или естествена история на заболяването, но е недостатъчен за изхвърляне на кетонни тела, получени от черния дроб, тъй като масивна хиперкетонемия възниква в условията на селективно увреждане на първичната окислителна съдба чрез загуба на функционални мутации до SCOT (Berry et al., 2012; Cotter et al., 2001).
Регулиране на HMGCS2 и SCOT/OXCT1
Отклонението на митохондриално от гена, кодиращ цитозолния HMGCS, се е случило в началото на еволюцията на гръбначните, поради необходимостта от подкрепа на чернодробната кетогенеза при видове с по-високо съотношение на мозъка към телесното тегло (Boukaftane et al., 1994; Cunnane и Crawford, 2003). Естествено срещащите се HMGCS2 мутации със загуба на функция при хора причиняват пристъпи на хипокетотична хипогликемия (Pitt et al., 2015; Thompson et al., 1997). Здравата експресия на HMGCS2 е ограничена до хепатоцитите и епитела на дебелото черво и нейната експресия и ензимната активност се координират чрез различни механизми (Mascaro et al., 1995; McGarry and Foster, 1980; Robinson and Williamson, 1980). Докато пълният обхват на физиологичните състояния, които влияят на HMGCS2, изисква допълнително изясняване, неговата експресия и/или активност се регулират по време на ранния постнатален период, стареене, диабет, гладуване или поглъщане на кетогенна диета (Balasse and Fery, 1989; Cahill GF Jr, 2006 ; Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). При плода метилирането на 5 фланкираща област на гена Hmgcs2 обратно корелира с неговата транскрипция и е частично обърнато след раждането (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al. ., 1983). По подобен начин, чернодробният Bdh1 проявява модел на експресия на развитие, нарастващ от раждането до отбиването и също се индуцира от кетогенна диета по начин, зависим от фибробластния растежен фактор (FGF)-21 (Badman et al., 2007; Zhang et al., 1989 ). Кетогенезата при бозайници е силно реагираща както на инсулин, така и на глюкагон, като съответно се потиска и стимулира (McGarry and Foster, 1977). Инсулинът потиска липолизата на мастната тъкан, като по този начин лишава кетогенезата от нейния субстрат, докато глюкагонът увеличава кетогенния поток чрез директен ефект върху черния дроб (Hegardt, 1999). Транскрипцията на Hmgcs2 се стимулира от транскрипционния фактор на вилицата FOXA2, който се инхибира чрез инсулин-фосфатидилинозитол-3-киназа/Akt и се индуцира от глюкагон-cAMP-p300 сигнализиране (Arias et al., 1995; Hegardt, 1999; , 1990; Thumelin et al., 1993; von Meyenn et al., 2013; Wolfrum et al., 2004; Wolfrum et al., 2003). PPAR? (Rodriguez et al., 1994) заедно с целта си, FGF21 (Badman et al., 2007) също индуцира транскрипция на Hmgcs2 в черния дроб по време на гладуване или прилагане на кетогенна диета (Badman et al., 2007; Inagaki et al., 2007 ). Индукция на PPAR? може да се случи преди прехода от фетална към неонатална физиология, докато активирането на FGF21 може да бъде благоприятно в ранния неонатален период чрез ?OHB-медиирано инхибиране на хистон деацетилаза (HDAC)-3 (Rando et al., 2016). mTORC1 (мишена на рапамицин комплекс 1 при бозайници) зависимо инхибиране на PPAR? транскрипционната активност също е ключов регулатор на генната експресия на Hmgcs2 (Sengupta et al., 2010), а чернодробният PER2, главен циркаден осцилатор, индиректно регулира експресията на Hmgcs2 (Chavan et al., 2016). Последните наблюдения показват, че екстрахепаталният тумор-индуциран интерлевкин-6 уврежда кетогенезата чрез PPAR? потискане (Flint et al., 2016).
HMGCS2 ензимната активност се регулира чрез множество PTM. Фосфорилирането на серин на HMGCS2 повишава неговата активност in vitro (Grimsrud et al., 2012). Активността на HMGCS2 се инхибира алостерично чрез сукцинилиране на сукцинил-CoA и лизинов остатък (Arias et al., 1995; Hegardt, 1999; Lowe and Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; 1975; Thumelin et al., 1993). Сукцинилирането на HMGCS2, HMGCL и BDH1 лизинови остатъци в чернодробните митохондрии са мишени на NAD+ зависимата деацилаза сиртуин 5 (SIRT5) (Rardin et al., 2013). Активността на HMGCS2 също се засилва от деацетилиране на SIRT3 лизин и е възможно кръстосаните смущения между ацетилирането и сукцинилирането да регулират активността на HMGCS2 (Rardin et al., 2013; Shimazu et al., 2013). Въпреки способността на тези PTM да регулират HMGCS2 Km и Vmax, флуктуациите на тези PTM все още не са внимателно картографирани и не са потвърдени като механистични двигатели на кетогенезата in vivo.
SCOT се експресира във всички клетки на бозайници, които съдържат митохондрии, с изключение на тези на хепатоцитите. Значението на активността на SCOT и кетолизата е демонстрирано при мишки SCOT-KO, които показват еднаква смъртност поради хиперкетонемична хипогликемия в рамките на 48 часа след раждането (Cotter et al., 2011). Тъканно-специфичната загуба на SCOT в неврони или скелетни миоцити предизвиква метаболитни аномалии по време на гладуване, но не е смъртоносна (Cotter et al., 2013b). При хората дефицитът на SCOT се проявява в началото на живота с тежка кетоацидоза, причиняваща летаргия, повръщане и кома (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al. , 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon и Cornblath, 1972). Относително малко се знае на клетъчно ниво за SCOT гена и регулаторите на експресия на протеини. Експресията на Oxct1 иРНК и SCOT протеинът и активността са намалени в кетозни състояния, вероятно чрез PPAR-зависими механизми (Fenselau and Wallis, 1974; Fenselau and Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al. ., 2001; Wentz et al., 2010). При диабетна кетоацидоза несъответствието между чернодробната кетогенеза и екстрахепаталното окисление се влошава от увреждане на активността на SCOT. Свръхекспресията на инсулин-независим глюкозен транспортер (GLUT1/SLC2A1) в кардиомиоцитите също инхибира експресията на Oxct1 ген и регулира надолу терминалното окисляване на кетоните в некетотично състояние (Yan et al., 2009). В черния дроб изобилието на Oxct1 иРНК се потиска от микроРНК-122 и метилиране на хистон H3K27me3, които са очевидни по време на прехода от фетален към неонатален период (Thorrez et al., 2011). Въпреки това, потискането на чернодробната експресия на Oxct1 в постнаталния период се дължи главно на евакуацията на Oxct1-експресиращи хематопоетични прогенитори от черния дроб, а не на загуба на съществуваща преди това експресия на Oxct1 в терминално диференцирани хепатоцити. Всъщност експресията на Oxct1 mRNA и SCOT протеин в диференцирани хепатоцити е изключително ниска (Orii et al., 2008).
SCOT също се регулира от PTM. Ензимът е хипер-ацетилиран в мозъците на SIRT3 KO мишки, които също показват намалено AcAc зависимо производство на ацетил-CoA (Dittenhafer-Reed et al., 2015). Неензимното нитриране на тирозинови остатъци на SCOT също отслабва неговата активност, което е докладвано в сърцата на различни модели на диабетни мишки (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). За разлика от това, нитрирането на триптофановите остатъци увеличава активността на SCOT (Br g re et al., 2010; Rebrin et al., 2007). Молекулни механизми на специфично за остатъци нитриране или денитриране, предназначени да модулират активността на SCOT, могат да съществуват и изискват изясняване.
Противоречия в екстрахепаталната кетогенеза
При бозайниците основният кетогенен орган е черният дроб и само хепатоцитите и чревните епителни клетки в изобилие експресират митохондриалната изоформа на HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry and Foster, 1980; Robinson1980 и Williamson) . Анаеробната бактериална ферментация на сложни полизахариди води до бутират, който се абсорбира от колоноцитите при бозайници за терминално окисление или кетогенеза (Cherbuy et al., 1995), което може да играе роля в диференциацията на колоноцитите (Wang et al., 2016). С изключение на чревните епителни клетки и хепатоцити, HMGCS2 почти липсва в почти всички други клетки на бозайници, но перспективата за екстрахепатална кетогенеза е повишена в туморните клетки, астроцитите на централната нервна система, бъбреците, панкреаса ? клетки, пигментен епител на ретината (RPE) и дори в скелетните мускули (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015 ; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). Ектопичен HMGCS2 е наблюдаван в тъкани, които нямат нетен кетогенен капацитет (Cook et al., 2016; Wentz et al., 2010), а HMGCS2 проявява проспективни независими от кетогенезата „моунлайтни“ активности, включително в рамките на клетъчното ядро (Chen et al. , 2016; Костюк и др., 2010; Меертенс и др., 1998).
Всяка екстрахепатална тъкан, която окислява кетонни тела, също има потенциал да натрупва кетонни тела чрез HMGCS2 независими механизми (фиг. 2А). Въпреки това, няма екстрахепатална тъкан, в която концентрацията на кетонни тела в стационарно състояние да надвишава тази в кръвообращението (Cotter et al., 2011; Cotter et al., 2013b; Harrison and Long, 1940), подчертавайки, че кетонните тела се транспортират надолу. концентрационен градиент чрез MCT1/2-зависими механизми. Един механизъм на очевидна екстрахепатална кетогенеза може всъщност да отразява относително увреждане на окислението на кетон. Допълнителни потенциални обяснения попадат в сферата на образуването на кетонни тела. Първо, de novo кетогенезата може да се случи чрез обратима ензимна активност на тиолаза и SCOT (Weidemann and Krebs, 1969). Когато концентрацията на ацетил-КоА е относително висока, реакциите, които обикновено са отговорни за окисляването на AcAc, протичат в обратна посока (GOLDMAN, 1954). Втори механизъм възниква, когато междинните продукти, получени от α-окисление, се натрупват поради затруднено място в цикъла на TCA, AcAc-CoA се превръща в l-?OHB-CoA чрез реакция, катализирана от митохондриална 3-хидроксиацил-CoA дехидрогеназа и допълнително от 3-хидроксибутирил CoA деацилаза до l-?OHB, която е неразличима чрез масспектрометрия или резонансна спектроскопия от физиологичния енантиомер d-?OHB (Reed and Ozand, 1980). l-?OHB може да бъде хроматографски или ензимно разграничен от d-?OHB и присъства в екстрахепаталните тъкани, но не и в черния дроб или кръвта (Hsu et al., 2011). Чернодробната кетогенеза произвежда само d-?OHB, единственият енантиомер, който е BDH субстрат (Ito et al., 1984; Lincoln et al., 1987; Reed and Ozand, 1980; Scofield et al., 1982; Scofield et al., 1982 г.). Трети HMGCS2-независим механизъм генерира d-?OHB чрез катаболизъм на аминокиселини, особено този на левцин и лизин. Четвъртият механизъм е само привиден, защото се дължи на артефакт на етикетиране и по този начин се нарича псевдокетогенеза. Това явление се дължи на обратимостта на реакциите на SCOT и тиолазата и може да причини надценяване на оборота на кетонните тела поради изотопното разреждане на маркера на кетонните тела в екстрахепаталната тъкан (Des Rosiers et al., 1990; Fink et al., 1988) . Независимо от това, псевдокетогенезата може да бъде незначителна в повечето контексти (Bailey et al., 1990; Keller et al., 1978). Схема (фиг. 2А) показва полезен подход, който да се приложи, като се има предвид повишената тъканна концентрация на кетони в стационарно състояние.


Наскоро бъбреците бяха обърнати внимание като потенциално кетогенен орган. В по-голямата част от щатите бъбрекът е нетен консуматор на кетонни тела, извлечени от черния дроб, отделяйки или реабсорбиращи кетонни тела от кръвния поток, а бъбреците обикновено не са генератор или концентратор на нетни кетонни тела (Robinson and Williamson, 1980). Авторите на класическо проучване стигат до заключението, че минималната бъбречна кетогенеза, определена количествено в изкуствена експериментална система, не е физиологично релевантна (Weidemann and Krebs, 1969). Напоследък се прави извод за бъбречна кетогенеза при миши модели с диабет и дефицит на аутофагия, но е по-вероятно многоорганните промени в метаболитната хомеостаза да променят интегративния кетогенен метаболизъм чрез въвеждане на множество органи (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). Една скорошна публикация предлага бъбречната кетогенеза като защитен механизъм срещу исхемично-реперфузионно увреждане в бъбреците (Tran et al., 2016). Абсолютните стационарни концентрации на ?OHB от екстракти от бъбречна тъкан на мишки са докладвани при ~4 mM. За да проверим дали това е приемливо, ние определихме количествено концентрациите на ?OHB в бъбречните екстракти от хранени и 12-часови гладни мишки. Серумните концентрации на ?OHB се увеличават от ~24 µM до 100 mM с 2-часово гладуване (фиг. 24B), докато концентрациите на βOHB в стационарно състояние в бъбреците се приближават до 2 µM в състояние на хранене и само 100 mM при 1-часово гладуване (фиг. 24C E), наблюдения, които са в съответствие с концентрациите, определени количествено преди повече от 2 години (Hems and Brosnan, 45). Остава възможно, че в кетозни състояния, кетонните тела, получени от черния дроб, биха могли да бъдат ренопротективни, но доказателствата за бъбречна кетогенеза изискват допълнително обосноваване. Убедителни доказателства, които подкрепят истинската екстрахепатална кетогенеза, бяха представени в RPE (Adijanto et al., 1970). Предполага се, че тази интригуваща метаболитна трансформация потенциално позволява на кетони, получени от RPE, да текат към фоторецепторните или клетките на глия на Mller, което може да помогне за регенерацията на външния сегмент на фоторецептора.
?OHB като сигнален посредник
Въпреки че са енергийно богати, кетонните тела упражняват провокативна „неканонична” сигнална роля в клетъчната хомеостаза (фиг. 3) (Newman and Verdin, 2014; Rojas-Morales et al., 2016). Например, ?OHB инхибира HDACs от клас I, което увеличава ацетилирането на хистони и по този начин индуцира експресията на гени, които ограничават оксидативния стрес (Shimazu et al., 2013). Самият ?OHB е хистонов ковалентен модификатор на лизинови остатъци в черния дроб на гладни или индуцирани от стрептозотоцин диабетни мишки (Xie et al., 2016) (вижте също по-долу, Интегрирането на метаболизма на кетонните тела, посттранслационна модификация и клетъчна физиология, и Кетонни тела, оксидативен стрес и невропротекция).

?OHB също е ефектор чрез G-протеин свързани рецептори. Чрез неясни молекулярни механизми, той потиска активността на симпатиковата нервна система и намалява общия разход на енергия и сърдечната честота чрез инхибиране на сигнализирането на късоверижни мастни киселини чрез G протеин-свързан рецептор 41 (GPR41) (Kimura et al., 2011). Един от най-изследваните сигнални ефекти на ?OHB протича чрез GPR109A (известен също като HCAR2), член на подсемейството на хидрокарбоксилната киселина GPCR, експресиран в мастната тъкан (бяла и кафява) (Tunaru et al., 2003) и в имунни клетки (Ahmed et al., 2009). ?OHB е единственият известен ендогенен лиганд на GPR109A рецептор (EC50 ~770 µM), активиран от d-?OHB, l-?OHB и бутират, но не и AcAc (Taggart et al., 2005). Високият праг на концентрация за активиране на GPR109A се постига чрез придържане към кетогенна диета, гладуване или по време на кетоацидоза, което води до инхибиране на липолизата на мастната тъкан. Антилиполитичният ефект на GPR109A протича чрез инхибиране на аденилциклазата и понижен сАМР, инхибирайки чувствителната към хормона триглицеридна липаза (Ahmed et al., 2009; Tunaru et al., 2003). Това създава отрицателна обратна връзка, в която кетозата поставя модулаторна спирачка на кетогенезата, като намалява освобождаването на неестерифицирани мастни киселини от адипоцитите (Ahmed et al., 2009; Taggart et al., 2005), ефект, който може да бъде уравновесен от симпатикусът, който стимулира липолизата. Ниацинът (витамин В3, никотинова киселина) е мощен (EC50 ~ 0.1 µM) лиганд за GRP109A, ефективно използван от десетилетия за дислипидемии (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al., 2010; Лукасова и др., 2011; Тунару и др., 2003). Докато ниацинът подобрява обратния транспорт на холестерол в макрофагите и намалява атеросклеротични лезии (Lukasova et al., 2011), ефектите на ?OHB върху атеросклеротични лезии остават неизвестни. Въпреки че GPR109A рецепторът изпълнява защитни роли и съществуват интригуващи връзки между употребата на кетогенна диета при инсулт и невродегенеративни заболявания (Fu et al., 2015; Rahman et al., 2014), защитната роля на ?OHB чрез GPR109A не е демонстрирана in vivo .
И накрая, ?OHB може да повлияе на апетита и ситост. Мета-анализ на проучвания, които измерват ефектите от кетогенните и много нискоенергийни диети, стига до заключението, че участниците, консумиращи тези диети, показват по-висока ситост в сравнение с контролните диети (Gibson et al., 2015). Въпреки това, правдоподобно обяснение за този ефект са допълнителните метаболитни или хормонални елементи, които могат да модулират апетита. Например, мишки, поддържани на кетогенна диета за гризачи, показват повишен разход на енергия в сравнение с мишки, хранени с контролно хранене, въпреки сходния калориен прием и циркулиращият лептин или гените на пептиди, регулиращи поведението на хранене, не са променени (Kennedy et al., 2007). Сред предложените механизми, които предполагат потискане на апетита от ?OHB, включва както сигнализиране, така и окисляване (Laeger et al., 2010). Хепатоцитно специфично заличаване на гена на циркадния ритъм (Per2) и изследванията за имунопреципитация на хроматин разкриват, че PER2 директно активира гена Cpt1a и индиректно регулира Hmgcs2, което води до нарушена кетоза при Per2 нокаут мишки (Chavan et al.,). Тези мишки показаха нарушено очакване на храна, което беше частично възстановено чрез системно приложение на ?OHB. Ще са необходими бъдещи изследвания, за да се потвърди централната нервна система като директна ?OHB мишена и дали е необходимо кетонно окисление за наблюдаваните ефекти или дали е включен друг сигнален механизъм. Други изследователи се позовават на възможността за локална кетогенеза, получена от астроцити във вентромедиалния хипоталамус като регулатор на приема на храна, но тези предварителни наблюдения също ще се възползват от генетични и базирани на потока оценки (Le Foll et al., 2016). Връзката между кетозата и недостига на хранителни вещества остава интерес, тъй като гладът и ситото са важни елементи при неуспешните опити за отслабване.
Интегриране на метаболизма на кетонното тяло, посттранслационна модификация и клетъчна физиология
Кетонните тела допринасят за разделени пулове от ацетил-КоА, ключов междинен продукт, който проявява важна роля в клетъчния метаболизъм (Pietrocola et al., 2015). Една роля на ацетил-CoA е да служи като субстрат за ацетилиране, ензимно катализирана хистонова ковалентна модификация (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016 ). Голям брой динамично ацетилирани митохондриални протеини, много от които могат да се появят чрез неензимни механизми, също се появиха от изчислителни протеомични изследвания (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013 ; Shimazu et al., 2010). Лизин деацетилазите използват цинков кофактор (напр. нуклеоцитозолни HDAC) или NAD+ като ко-субстрат (сиртуини, SIRT) (Choudhary et al., 2014; Menzies et al., 2016). Ацетилпротеомът служи както като сензор, така и като ефектор на общия клетъчен ацетил-КоА пул, тъй като физиологичните и генетичните манипулации водят до неензимни глобални вариации на ацетилирането (Weinert et al., 2014). Тъй като вътреклетъчните метаболити служат като модулатори на ацетилирането на лизинови остатъци, важно е да се вземе предвид ролята на кетонните тела, чието изобилие е силно динамично.
?OHB е епигенетичен модификатор чрез поне два механизма. Повишените нива на ?OHB, предизвикани от гладуване, ограничаване на калориите, директно приложение или продължително упражнение, провокират инхибиране на HDAC или активиране на хистон ацетилтрансфераза (Marosi et al., 2016; Sleiman et al., 2016) или до оксидативен стрес (Shimazu et al.), 2013 3. . Инхибирането на ?OHB на HDAC2016 може да регулира метаболитната физиология на новороденото (Rando et al., 2016). Независимо, самият ?OHB директно модифицира остатъците от хистон лизин (Xie et al., XNUMX). Продължителното гладуване или предизвиканата от стептозотоцин диабетна кетоацидоза повишава хистонова α-хидроксибутирилация. Въпреки че броят на местата на лизин а-хидроксибутирилиране и ацетилиране е сравним, се наблюдава стехиометрично по-голямо хистоново р-хидроксибутирилиране, отколкото ацетилиране. Различни гени са повлияни от хистон лизин а-хидроксибутирилиране, срещу ацетилиране или метилиране, което предполага различни клетъчни функции. Дали а-хидроксибутирилирането е спонтанно или ензимно, не е известно, но разширява обхвата от механизми чрез кетонни тела, които динамично влияят на транскрипцията.
Събитията на съществено клетъчно препрограмиране по време на ограничаване на калориите и липса на хранителни вещества могат да бъдат медиирани в SIRT3- и SIRT5-зависимо митохондриално деацетилиране и десукцинилиране, съответно, регулирайки кетогенните и кетолитичните протеини на пост-транслационно ниво в черния дроб и екстрахепаталните тъкани на ReDittenhafer, 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Въпреки че стехиометричното сравнение на заетите места не е непременно свързано директно с промените в метаболитния поток, митохондриалното ацетилиране е динамично и може да се задвижва от концентрацията на ацетил-КоА или митохондриалното рН, а не от ензимните ацетилтрансферази (Wagner and Payne, 2013). Това, че SIRT3 и SIRT5 модулират активността на ензимите, метаболизиращи кетонното тяло, провокира въпроса за реципрочната роля на кетоните при извайването на ацетилпротеома, сукцинилпротеома и други динамични клетъчни цели. Всъщност, тъй като вариациите на кетогенезата отразяват концентрациите на NAD+, производството и изобилието на кетони могат да регулират активността на сиртуин, като по този начин повлияят на общите пулове ацетил-CoA/сукцинил-CoA, ацилпротеома и по този начин митохондриалната и клетъчната физиология. а-хидроксибутирилирането на остатъци от ензим лизин може да добави още един слой към клетъчното препрограмиране. В екстрахепаталните тъкани окисляването на кетонните тела може да стимулира аналогични промени в клетъчната хомеостаза. Докато компартментацията на пуловете на ацетил-CoA е силно регулирана и координира широк спектър от клетъчни промени, способността на кетонните тела да оформят директно концентрациите на ацетил-CoA в митохондриите и цитоплазмата изисква изясняване (Chen et al., 2012; Corbet et al., 2016; Pougovkina et al., 2014; Schwer et al., 2009; Wellen and Thompson, 2012). Тъй като концентрациите на ацетил-КоА са строго регулирани, а ацетил-КоА е непроницаем за мембраната, от решаващо значение е да се вземат предвид двигателните механизми, координиращи хомеостазата на ацетил-КоА, включително скоростта на производство и терминално окисление в TCA цикъла, превръщане в кетонни тела, митохондриални изтичане чрез карнитин ацетилтрансфераза (CrAT) или износ на ацетил-КоА в цитозола след превръщане в цитрат и освобождаване от ATP цитрат лиаза (ACLY). Ключовите роли на тези последни механизми в клетъчния ацетилпротеом и хомеостазата изискват съвпадащо разбиране на ролите на кетогенезата и окислението на кетоните (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen и Thompson, 2012). Ще са необходими конвергентни технологии в метаболомиката и ацилпротеомиката в условията на генетично манипулирани модели, за да се определят цели и резултати.
Противовъзпалителни и противовъзпалителни реакции към кетонните тела
Кетозата и кетонните тела модулират възпалението и функцията на имунните клетки, но са предложени различни и дори несъответстващи механизми. Продължителното недостиг на хранителни вещества намалява възпалението (Youm et al., 2015), но хроничната кетоза на диабет тип 1 е провъзпалително състояние (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012 ). Основаните на механизма сигнални роли за ?OHB при възпаление се появяват, защото много клетки на имунната система, включително макрофаги или моноцити, обилно експресират GPR109A. Докато ?OHB упражнява предимно противовъзпалителен отговор (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), високи концентрации на кетонни тела, особено AcAc, могат предизвикват провъзпалителен отговор (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012).
Противовъзпалителните роли на GPR109A лигандите при атеросклероза, затлъстяване, възпалително заболяване на червата, неврологични заболявания и рак са разгледани (Graff et al., 2016). Експресията на GPR109A се увеличава в RPE клетки на диабетни модели, пациенти с диабет при хора (Gambhir et al., 2012) и в микроглия по време на невродегенерация (Fu et al., 2014). Противовъзпалителните ефекти на ?OHB се засилват от свръхекспресията на GPR109A в клетките на RPE и се отменят чрез фармакологично инхибиране или генетично нокаутиране на GPR109A (Gambhir et al., 2012). ?OHB и екзогенна никотинова киселина (Taggart et al., 2005), и двете придават противовъзпалителни ефекти в TNF? или LPS-индуцирано възпаление чрез намаляване на нивата на провъзпалителни протеини (iNOS, COX-2) или секретирани цитокини (TNFa, IL-1a, IL-6, CCL2/MCP-1), отчасти чрез инхибиране на NF -?B транслокация (Fu et al., 2014; Gambhir et al., 2012). ?OHB намалява ER стреса и NLRP3 инфламазомата, активирайки реакцията на антиоксидантен стрес (Bae et al., 2016; Youm et al., 2015). Въпреки това, при невродегенеративно възпаление, GPR109A-зависимата ?OHB-медиирана защита не включва възпалителни медиатори като сигнализиране на пътя на MAPK (напр. ERK, JNK, p38) (Fu et al., 2014), но може да изисква COX-1-зависим PGD2 производство (Rahman et al., 2014). Интригуващо е, че макрофагът GPR109A е необходим за упражняване на невропротективен ефект в модел на исхемичен инсулт (Rahman et al., 2014), но способността на ?OHB да инхибира възпалението на NLRP3 в макрофагите, получени от костния мозък, е независима от GPR109A ., 2015 г.). Въпреки че повечето проучвания свързват ?OHB с противовъзпалителни ефекти, ?OHB може да е провъзпалително и да повишава маркерите на липидна пероксидация в хепатоцитите на телетата (Shi et al., 2014). По този начин противовъзпалителният ефект на αOHB може да зависи от типа на клетката, концентрацията на αOHB, продължителността на експозиция и наличието или отсъствието на ко-модулатори.
За разлика от ?OHB, AcAc може да активира про-възпалителна сигнализация. Повишеният AcAc, особено с висока концентрация на глюкоза, засилва увреждането на ендотелните клетки чрез механизъм, зависим от NADPH оксидаза/оксидативен стрес (Kanikarla-Marie and Jain, 2015). Високите концентрации на AcAc в пъпната връв на майки с диабет са свързани с по-висока скорост на окисление на протеини и концентрация на MCP-1 (Kurepa et al., 2012). Високият AcAc при пациенти с диабет е свързан с TNF? експресия (Jain et al., 2002) и AcAc, но не ?OHB, индуцира TNF?, MCP-1 експресия, натрупване на ROS и намалено ниво на cAMP в U937 човешки моноцитни клетки (Jain et al., 2002; Kurepa et al. ., 2012 г.).
Сигналните явления, зависими от кетонното тяло, често се задействат само при високи концентрации на кетонно тяло (> 5 mM) и в случай на много проучвания, свързващи кетоните с про- или противовъзпалителни ефекти, чрез неясни механизми. В допълнение, поради противоречивите ефекти на ?OHB срещу AcAc върху възпалението и способността на съотношението AcAc/?OHB да повлияе на митохондриалния редокс потенциал, най-добрите експерименти, оценяващи ролите на кетонните тела върху клетъчните фенотипове, сравняват ефектите на AcAc и ? OHB в различни съотношения и при различни кумулативни концентрации [напр. (Saito et al., 2016)]. И накрая, AcAc може да бъде закупен в търговската мрежа само като литиева сол или като етилов естер, който изисква основна хидролиза преди употреба. Литиевият катион независимо индуцира каскади на сигнална трансдукция (Manji et al., 1995), а AcAc анионът е лабилен. И накрая, изследванията, използващи рацемичен d/l-?OHB, могат да бъдат объркани, тъй като само d-?OHB стереоизомерът може да бъде окислен до AcAc, но d-?OHB и l-?OHB могат всеки да сигнализира чрез GPR109A, да инхибира NLRP3 инфламазомата, и служат като липогенни субстрати.
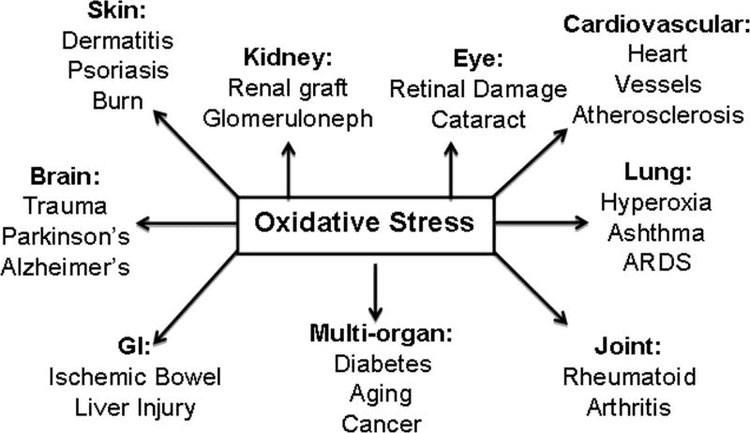
Кетонни тела, оксидативен стрес и неврозащита
Оксидативният стрес обикновено се дефинира като състояние, в което ROS са представени в излишък, поради прекомерно производство и/или нарушено елиминиране. Ролите на кетонните тела за смекчаване на антиоксидантния и оксидативния стрес са широко описани както in vitro, така и in vivo, особено в контекста на невропротекцията. Тъй като повечето неврони не генерират ефективно високоенергийни фосфати от мастни киселини, но окисляват кетонните тела, когато въглехидратите са в недостиг, невропротективните ефекти на кетонните тела са особено важни (Cahill GF Jr, 2006; Edmond et al., 1987; Yang et al., 1987). В моделите на оксидативен стрес индукцията на BDH1 и потискането на SCOT предполагат, че метаболизмът на кетоновите тела може да бъде препрограмиран, за да поддържа разнообразна клетъчна сигнализация, редокс потенциал или метаболитни изисквания (Nagao et al., 2016; Tieu et al., 2003).
Кетонните тела намаляват степента на клетъчно увреждане, нараняване, смърт и по-ниска апоптоза в неврони и кардиомиоцити (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). Извикваните механизми са разнообразни и не винаги са линейно свързани с концентрацията. Ниските милимоларни концентрации на (d или l)-?OHB улавят ROS (хидроксил анион), докато AcAc улавя множество видове ROS, но само при концентрации, които надхвърлят физиологичния диапазон (IC50 20 mM) (Haces et al., 67) . Обратно, благоприятното влияние върху окислително-редукционния потенциал на веригата за транспортиране на електрони е механизъм, обикновено свързан с d-?OHB. Докато и трите кетонни тела (d/l-?OHB и AcAc) намаляват смъртта на невронните клетки и натрупването на ROS, предизвикано от химическо инхибиране на гликолизата, само d-?OHB и AcAc предотвратяват намаляването на невроналния АТФ. Обратно, в хипогликемичен in vivo модел, (d или l)-?OHB, но не и AcAc предотвратяват хипокампалната липидна пероксидация (Haces et al., 2008; Maalouf et al., 2008; Marosi et al., 2007; Murphy, 2016 ; Tieu et al., 2009). Изследвания in vivo на мишки, хранени с кетогенна диета (2003% kcal мазнини и 87% протеин) показват невроанатомични вариации на антиоксидантния капацитет (Ziegler et al., 13), където най-дълбоките промени се наблюдават в хипокампуса, с повишаване на глутатион пероксидазата и общата антиоксидантни способности.
Кетогенната диета, кетонните естери (вижте също Терапевтично използване на кетогенна диета и екзогенни кетонни тела) или прилагането на ?OHB оказват невропротекция при модели на исхемичен инсулт (Rahman et al., 2014); болест на Паркинсон (Tieu et al., 2003); припадък на кислородна токсичност на централната нервна система (D'Agostino et al., 2013); епилептични спазми (Yum et al., 2015); синдром на митохондриална енцефаломиопатия, лактатна ацидоза и подобни на инсулт (MELAS) епизоди (Frey et al., 2016) и болест на Алцхаймер (Cunnane and Crawford, 2003; Yin et al., 2016). Обратно, скорошен доклад демонстрира хистопатологични доказателства за невродегенеративна прогресия чрез кетогенна диета в модел на трансгенна мишка на анормално възстановяване на митохондриална ДНК, въпреки увеличаването на митохондриалната биогенеза и антиоксидантните сигнатури (Lauritzen et al., 2016). Други противоречиви доклади предполагат, че излагането на високи концентрации на кетонни тела предизвиква оксидативен стрес. Високите дози ?OHB или AcAc индуцират секреция на азотен оксид, липидна пероксидация, намалена експресия на SOD, глутатион пероксидаза и каталаза в хепатоцитите на телета, докато в хепатоцитите на плъхове индукцията на MAPK пътя се приписва на AcAc, но не и на ?OHB (Abdelmegeed et al. ; Shi et al., 2004; Shi et al., 2014).
Взети заедно, повечето доклади свързват ?OHB с отслабването на оксидативния стрес, тъй като приложението му инхибира производството на ROS/супероксид, предотвратява липидна пероксидация и окисляване на протеини, повишава нивата на антиоксидантния протеин и подобрява митохондриалното дишане и производството на АТФ (Abdelmegeed et al., 2004); Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Maalouf et al., 2007; Maalouf и Rho, 2008; Marosi et al., 2016; Tieu et al., 2003; Yin et al., 2016; Ziegler et al., 2003). Докато AcAc е по-пряко свързан от ?OHB с индуцирането на оксидативен стрес, тези ефекти не винаги се разграничават лесно от проспективните провъзпалителни отговори (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kanikarla-Marie и Джайн, 2016 г.). Освен това е от решаващо значение да се има предвид, че очевидната антиоксидантна полза, предоставена от плейотропните кетогенни диети, може да не бъде трансдуцирана от самите кетонни тела, а невропротекцията, предоставена от кетонните тела, може да не се дължи изцяло на оксидативния стрес. Например по време на лишаване от глюкоза, в модел на лишаване от глюкоза в кортикалните неврони, ?OHB стимулира автофагичния поток и предотвратява натрупването на автофагозома, което е свързано с намалена невронна смърт (Camberos-Luna et al., 2016). d-?OHB индуцира също и каноничните антиоксидантни протеини FOXO3a, SOD, MnSOD и каталаза, проспективно чрез HDAC инхибиране (Nagao et al., 2016; Shimazu et al., 2013).
Безалкохолна мастна чернодробна болест (NAFLD) и метаболизъм на кетонното тяло
Свързаната със затлъстяването NAFLD и неалкохолен стеатохепатит (NASH) са най-честите причини за чернодробно заболяване в западните страни (Rinella and Sanyal, 2016), а индуцираната от NASH чернодробна недостатъчност е една от най-честите причини за чернодробна трансплантация. Докато излишното съхранение на триацилглицероли в хепатоцити >5% от теглото на черния дроб (NAFL) само по себе си не причинява дегенеративна чернодробна функция, прогресията към NAFLD при хората корелира със системна инсулинова резистентност и повишен риск от диабет тип 2 и може да допринесе за патогенезата на сърдечно-съдови заболявания и хронично бъбречно заболяване (Fabbrini et al., 2009; Targher et al., 2010; Targher and Byrne, 2013). Патогенните механизми на NAFLD и NASH не са напълно разбрани, но включват аномалии на хепатоцитния метаболизъм, хепатоцитната аутофагия и стрес на ендоплазмения ретикулум, функцията на чернодробните имунни клетки, възпаление на мастната тъкан и системни възпалителни медиатори (Fabbrini et al., 2009, Masaniu, 2013; ; Targher et al., 2010; Yang et al., 2010). Смущенията в метаболизма на въглехидратите, липидите и аминокиселините се появяват и допринасят за затлъстяване, диабет и NAFLD при хора и в моделни организми [прегледани в (Farese et al., 2012; Lin and Accili, 2011; Newgard, 2012; Samuel и Шулман, 2012; Слънце и Лазар, 2013)]. Докато хепатоцитните аномалии в метаболизма на цитоплазмените липиди често се наблюдават при NAFLD (Fabbrini et al., 2010b), ролята на митохондриалния метаболизъм, който управлява окислителното изхвърляне на мазнините, е по-малко ясна в патогенезата на NAFLD. Аномалии на митохондриалния метаболизъм се появяват и допринасят за патогенезата на NAFLD/NASH (Hyotylainen et al., 2016; Serviddio et al., 2011; Serviddio et al., 2008; Wei et al., 2008). Има общо (Felig et al., 1974; Iozzo et al., 2010; Koliaki et al., 2015; Satapati et al., 2015; Satapati et al., 2012; Sunny et al., 2011), но не е еднородно ( Koliaki и Roden, 2013; Perry et al., 2016; Rector et al., 2010) консенсус, че преди развитието на добросъвестния NASH, чернодробното митохондриално окисление и по-специално окисляването на мазнините се увеличава при затлъстяване, системна инсулинова резистентност и NAFLD. Вероятно е, че с напредването на NAFLD се появява хетерогенност на окислителния капацитет, дори сред отделните митохондрии, и в крайна сметка оксидативната функция се нарушава (Koliaki et al., 2015; Rector et al., 2010; Satapati et al., 2008; Satapati et al. ., 2012 г.).
Кетогенезата често се използва като заместител на окисляването на чернодробните мазнини. Нарушенията на кетогенезата се появяват с напредването на NAFLD при животински модели и вероятно при хора. Чрез непълно дефинирани механизми, хиперинсулинемията потиска кетогенезата, вероятно допринасяйки за хипокетонемия в сравнение с слабите контроли (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters2009 et al., , ; , 2011; Vice et al., 2005). Независимо от това, способността на концентрациите на циркулиращи кетонни тела да предскажат NAFLD е противоречива (M nnist et al., 2015; Sanyal et al., 2001). Стабилните количествени магнитно-резонансни спектроскопски методи в животински модели разкриват повишена скорост на обмяна на кетони с умерена инсулинова резистентност, но намалените нива са очевидни при по-тежка инсулинова резистентност (Satapati et al., 2012; Sunny et al., 2010). При затлъстели хора с мастен черен дроб кетогенната скорост е нормална (Bickerton et al., 2008; Sunny et al., 2011) и следователно скоростта на кетогенезата е намалена спрямо увеличеното натоварване на мастните киселини в хепатоцитите. Следователно, ацетил-КоА, извлечен от α-окисление, може да бъде насочен към терминално окисление в TCA цикъла, увеличавайки терминалното окисление, глюконеогенеза, задвижвана от фосфоенолпируват чрез анаплероза/катаплероза и оксидативен стрес. Ацетил-КоА също вероятно претърпява износ от митохондриите като цитрат, прекурсорен субстрат за липогенезата (фиг. 4) (Satapati et al., 2015; Satapati et al., 2012; Solinas et al., 2015). Докато кетогенезата става по-малко чувствителна към инсулин или гладуване с продължително затлъстяване (Satapati et al., 2012), основните механизми и последиците от това надолу по веригата остават ненапълно разбрани. Последните данни показват, че mTORC1 потиска кетогенезата по начин, който може да бъде след инсулиновата сигнализация (Kucejova et al., 2016), което е в съответствие с наблюденията, че mTORC1 инхибира PPAR?-медиирана индукция на Hmgcs2 (Sengupta et al., 2010). също вижте Регламент на HMGCS2 и SCOT/OXCT1).

Предварителните наблюдения от нашата група предполагат неблагоприятни чернодробни последици от кетогенната недостатъчност (Cotter et al., 2014). За да проверим хипотезата, че нарушената кетогенеза, дори в състояния, наситени с въглехидрати и по този начин „некетогенни“ състояния, допринася за анормален метаболизъм на глюкозата и провокира стеатохепатит, ние генерирахме миши модел на изразена кетогенна недостатъчност чрез прилагане на антисенс олигонукле (ASO) Hmgcs2. Загубата на HMGCS2 при възрастни мишки, хранени с ниско съдържание на мазнини, причинява лека хипергликемия и значително повишено производство на стотици чернодробни метаболити, набор от които силно предполага активиране на липогенезата. Диетично хранене с високо съдържание на мазнини на мишки с недостатъчна кетогенеза води до обширно увреждане и възпаление на хепатоцитите. Тези констатации подкрепят централните хипотези, че (i) кетогенезата не е пасивен път на преливане, а по-скоро динамичен възел в чернодробната и интегрирана физиологична хомеостаза, и (ii) разумното кетогенно увеличаване за смекчаване на NAFLD/NASH и нарушен метаболизъм на чернодробната глюкоза е достойно за изследване .
Как може нарушената кетогенеза да допринесе за чернодробно увреждане и променена глюкозна хомеостаза? Първото съображение е дали виновникът е дефицитът на кетогенен поток или самите кетони. Неотдавнашен доклад предполага, че кетонните тела могат да смекчат индуцираното от оксидативен стрес чернодробно увреждане в отговор на n-3 полиненаситени мастни киселини (Pawlak et al., 2015). Припомнете си, че поради липса на експресия на SCOT в хепатоцитите, кетонните тела не се окисляват, но те могат да допринесат за липогенезата и да служат на различни сигнални роли, независимо от тяхното окисление (вижте също неокислителни метаболитни съдби на кетонни тела и ?OHB като сигнален медиатор). Възможно е също така получените от хепатоцити кетонни тела да служат като сигнал и/или метаболит за съседни типове клетки в чернодробния ацинус, включително звездни клетки и макрофаги на Купферова клетка. Докато ограничената налична литература предполага, че макрофагите не са в състояние да окисляват кетонни тела, това е измерено само с помощта на класически методологии и само в перитонеални макрофаги (Newsholme et al., 1986; Newsholme et al., 1987), което показва, че повторното оценката е подходяща, като се има предвид изобилната експресия на SCOT в макрофаги, получени от костен мозък (Youm et al., 2015).
Хепатоцитният кетогенен поток може също да бъде цитопротективен. Докато оздравителните механизми може да не зависят от кетогенезата сама по себе си, кетогенните диети с ниско съдържание на въглехидрати са свързани с подобряване на NAFLD (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar and Crawford, 2012) . Нашите наблюдения показват, че кетогенезата на хепатоцитите може да даде обратна връзка и да регулира потока на TCA цикъла, анаплеротичния поток, глюконеогенезата, получена от фосфоенолпируват (Cotter et al., 2014) и дори оборота на гликоген. Кетогенното увреждане насочва ацетил-КоА към увеличаване на потока на TCA, който в черния дроб е свързан с повишено ROS-медиирано увреждане (Satapati et al., 2015; Satapati et al., 2012); принуждава отклоняване на въглерод в de novo синтезирани липидни видове, които могат да се окажат цитотоксични; и предотвратява повторното окисление на NADH до NAD+ (Cotter et al., 2014) (фиг. 4). Взети заедно, бъдещи експерименти са необходими за справяне с механизмите, чрез които относителната кетогенна недостатъчност може да стане неадаптивна, да допринесе за хипергликемия, да провокира стеатохепатит и дали тези механизми функционират при човешки NAFLD/NASH. Тъй като епидемиологичните данни предполагат нарушена кетогенеза по време на прогресията на стеатохепатит (Embade et al., 2016; Marinou et al., 2011; M nnist et al., 2015; Pramfalk et al., 2015; Safaei et al., 2016) терапиите, които повишават чернодробната кетогенеза, могат да се окажат полезни (Degirolamo et al., 2016; Honda et al., 2016).
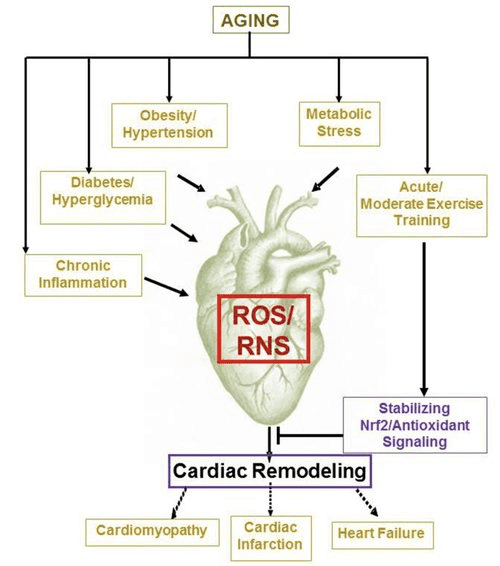
Кетонни тела и сърдечна недостатъчност (HF)
С метаболитна скорост над 400 kcal/kg/ден и оборот от 6 kg ATP/ден, сърцето е органът с най-висок разход на енергия и окислително търсене (Ashrafian et al., 35; Wang et al., 2007б). По-голямата част от енергийния оборот на миокарда се намира в митохондриите, а 2010% от това снабдяване произхожда от ФАО. Сърцето е всеядно и гъвкаво при нормални условия, но патологично ремоделиращото се сърце (например поради хипертония или инфаркт на миокарда) и диабетното сърце стават метаболитно негъвкави (Balasse and Fery, 70; BING, 1989; Fukao et al., 1954 ; Lopaschuk et al., 2004; Taegtmeyer et al., 2010; Taegtmeyer et al., 1980; Young et al., 2002). Всъщност генетично програмираните аномалии на сърдечния горивен метаболизъм при миши модели провокират кардиомиопатия (Carley et al., 2002; Neubauer, 2014). При физиологични условия нормалните сърца окисляват кетонните тела пропорционално на доставянето им, за сметка на окисляването на мастни киселини и глюкоза, а миокардът е най-големият консуматор на кетонно тяло на единица маса (BING, 2007; Crawford et al., 1954; GARLAND et al. ., 2009; Hasselbaink et al., 1962; Jeffrey et al., 2003; Pelletier et al., 1995; Tardif et al., 2007; Yan et al., 2001). В сравнение с окисляването на мастни киселини, кетонните тела са по-енергийно ефективни, давайки повече енергия, налична за синтеза на АТФ на молекула вложен кислород (P/O съотношение) (Kashiwaya et al., 2009; Sato et al., 2010; Veech, 1995) . Окислението на кетонните тела също води до потенциално по-висока енергия от FAO, поддържайки окисление на убихинона, което увеличава редокс обхвата във веригата за транспорт на електрони и прави повече енергия налична за синтезиране на ATP (Sato et al., 2004; Veech, 1995). Окислението на кетонните тела може също да намали производството на ROS и по този начин оксидативния стрес (Veech, 2004).
Предварителните интервенционни и наблюдателни проучвания показват потенциална оздравителна роля на кетонните тела в сърцето. В контекста на експериментална исхемия/реперфузионно увреждане, кетонните тела придават потенциални кардиопротективни ефекти (Al-Zaid et al., 2007; Wang et al., 2008), вероятно поради увеличаването на изобилието на митохондриите в сърцето или повишаването на регулирането на решаващо окислително фосфорилиране медиатори (Snorek et al., 2012; Zou et al., 2002). Последните проучвания показват, че използването на кетонни тела се увеличава при неуспешни сърца на мишки (Aubert et al., 2016) и хора (Bedi et al., 2016), подкрепяйки предишни наблюдения при хора (BING, 1954; Fukao et al., 2000; Janardhan et al., 2011; Longo et al., 2004; Rudolph and Schinz, 1973; Tildon и Cornblath, 1972). Концентрациите на циркулиращите кетонни тела се повишават при пациенти със сърдечна недостатъчност, право пропорционално на налягането на пълнене, наблюдения, чийто механизъм и значение остават неизвестни (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al. ., 1972), но мишките със селективен дефицит на SCOT в кардиомиоцитите показват ускорено патологично камерно ремоделиране и ROS сигнатури в отговор на хирургично индуцирано увреждане от претоварване с налягане (Schugar et al., 2014).
Последните интригуващи наблюдения в терапията на диабета разкриха потенциална връзка между метаболизма на миокарда на кетони и патологичното камерно ремоделиране (Фиг. 5). Инхибирането на бъбречния проксимален тубулен натриев/глюкозен ко-транспортер 2 (SGLT2i) повишава концентрациите на циркулиращи кетонни тела при хора (Ferrannini et al., 2016a; Inagaki et al., 2015) и мишки (Suzuki et al., 2014) чернодробна кетогенеза (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz и Leiter, 2015; Mudaliar et al., 2015). Поразително е, че поне един от тези агенти намалява хоспитализацията при сърдечна недостатъчност (напр., както е разкрито от проучването EMPA-REG OUTOME) и подобрява сърдечно-съдовата смъртност (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a ; Zinman et al., 2015). Докато движещите механизми зад благоприятните резултати от СН за свързания SGLT2i остават активно обсъждани, ползата от оцеляването вероятно е многофакторна, проспективно включваща кетоза, но също и благотворни ефекти върху теглото, кръвното налягане, нивата на глюкоза и пикочна киселина, артериалната скованост, симпатиковата нервна система, осмотичното диуреза/намален плазмен обем и повишен хематокрит (Raz and Cahn, 2016; Vallon and Thomson, 2016). Взети заедно, схващането, че терапевтично увеличаващата се кетонемия при пациенти със СН или при тези с висок риск от развитие на СН, остава противоречива, но е в процес на активно изследване в предклинични и клинични проучвания (Ferrannini et al., 2016b; Kolwicz et al., 2016; Lopaschuk и Verma, 2016; Mudaliar et al., 2016; Taegtmeyer, 2016).

Кетонни тела в биологията на рака
Връзките между кетонните тела и рака бързо се появяват, но проучванията както върху животински модели, така и при хора са довели до различни заключения. Тъй като кетонният метаболизъм е динамичен и реагира на хранителните състояния, примамливо е да се търсят биологични връзки с рака поради потенциала за прецизно ръководени хранителни терапии. Раковите клетки се подлагат на метаболитно препрограмиране, за да поддържат бърза клетъчна пролиферация и растеж (DeNicola and Cantley, 2015; Pavlova and Thompson, 2016). Класическият ефект на Варбург в метаболизма на раковите клетки възниква от доминиращата роля на гликолизата и ферментацията на млечна киселина за пренос на енергия и компенсиране на по-ниската зависимост от окислителното фосфорилиране и ограниченото митохондриално дишане (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Глюкозният въглерод се насочва основно чрез гликолиза, пентозофосфатен път и липогенеза, които заедно осигуряват междинни продукти, необходими за разширяване на туморната биомаса (Grabbacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). Адаптирането на раковите клетки към лишаване от глюкоза се осъществява чрез способността да се използват алтернативни източници на гориво, включително ацетат, глутамин и аспартат (Jaworski et al., 2016; Sullivan et al., 2015). Например, ограничен достъп до пируват разкрива способността на раковите клетки да превръщат глутамин в ацетил-КоА чрез карбоксилиране, поддържайки както енергийните, така и анаболните нужди (Yang et al., 2014). Интересна адаптация на раковите клетки е използването на ацетат като гориво (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright and Simone, 2016; Yoshii et al., 2015). Ацетатът също е субстрат за липогенезата, която е от решаващо значение за пролиферацията на туморни клетки, а усилването на този липогенен канал е свързано с по-кратка преживяемост на пациентите и по-голямо туморно натоварване (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al. ., 2015 г.).
Нераковите клетки лесно прехвърлят своя енергиен източник от глюкоза към кетонни тела по време на лишаване от глюкоза. Тази пластичност може да е по-променлива при видовете ракови клетки, но in vivo имплантирани мозъчни тумори окисляват [2,4-13C2]-?OHB до подобна степен като заобикалящата мозъчна тъкан (De Feyter et al., 2016). Моделите на „обратен ефект на Варбург“ или „туморен метаболизъм с две отделения“ предполагат, че раковите клетки индуцират производството на ?OHB в съседни фибробласти, осигурявайки енергийните нужди на туморните клетки (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012) . В черния дроб изместването на хепатоцитите от кетогенеза към окисление на кетони в клетките на хепатоцелуларен карцином (хепатом) е в съответствие с активирането на BDH1 и SCOT активностите, наблюдавани в две хепатомни клетъчни линии (Zhang et al., 1989). Наистина, хепатомните клетки експресират OXCT1 и BDH1 и окисляват кетони, но само когато серумът е гладен (Huang et al., 2016). Като алтернатива е предложена и кетогенеза на туморни клетки. Динамичните промени в експресията на кетогенния ген се проявяват по време на ракова трансформация на епитела на дебелото черво, клетъчен тип, който нормално експресира HMGCS2, а скорошен доклад предполага, че HMGCS2 може да бъде прогностичен маркер за лоша прогноза при колоректални и плоскоклетъчни карциноми (Camarero et al. 2006; Чен и др., 2016). Остава да се определи дали тази асоциация изисква или включва кетогенеза, или съвместна функция на HMGCS2. Обратно, очевидното производство на ?OHB от клетките на меланома и глиобластома, стимулирано от PPAR? агонист фенофибрат, се свързва със спиране на растежа (Grabacka et al., 2016). Необходими са по-нататъшни проучвания, за да се характеризират ролите на експресията на HMGCS2/SCOT, кетогенезата и окисляването на кетони в раковите клетки.
Отвъд сферата на горивния метаболизъм, кетоните наскоро бяха замесени в биологията на раковите клетки чрез сигнален механизъм. Анализът на BRAF-V600E+ меланом показва OCT1-зависима индукция на HMGCL по онкогенен BRAF-зависим начин (Kang et al., 2015). Увеличаването на HMGCL е свързано с по-висока клетъчна концентрация на AcAc, което от своя страна засилва взаимодействието на BRAFV600E-MEK1, усилвайки MEK-ERK сигнализирането в цикъл за предаване, който задвижва пролиферацията и растежа на туморните клетки. Тези наблюдения повдигат интригуващия въпрос за проспективната екстрахепатална кетогенеза, която след това поддържа сигнален механизъм (вижте също ?OHB като сигнален медиатор и Противоречия в екстрахепаталната кетогенеза). Също така е важно да се вземат предвид независимите ефекти на AcAc, d-?OHB и l-?OHB върху метаболизма на рака, а когато се разглежда HMGCL, катаболизмът на левцина също може да бъде нарушен.
Ефектите от кетогенните диети (вижте също Терапевтично използване на кетогенна диета и екзогенни кетонни тела) при ракови животински модели са разнообразни (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al. ., 2014; Seyfried et al., 2011; Shukla et al., 2014). Докато епидемиологичните асоциации между затлъстяването, рака и кетогенните диети се обсъждат (Liskiewicz et al., 2016; Wright and Simone, 2016), мета-анализ, използващ кетогенни диети в животински модели и в проучвания при хора, предполага благотворно въздействие върху оцеляването, с ползи, проспективно свързани с големината на кетозата, времето на започване на диетата и местоположението на тумора (Klement et al., 2016; Woolf et al., 2016). Лечението на ракови клетки на панкреаса с кетонни тела (d-?OHB или AcAc) инхибира растежа, пролиферацията и гликолизата, а кетогенната диета (81% kcal мазнини, 18% протеин, 1% въглехидрати) намалява in vivo теглото на тумора, гликемията и повишено мускулно и телесно тегло при животни с имплантиран рак (Shukla et al., 2014). Подобни резултати са наблюдавани при използване на метастатичен клетъчен модел на глиобластом при мишки, които получават кетонни добавки в диетата (Poff et al., 2014). Обратно, кетогенната диета (91% kcal мазнини, 9% протеин) повишава циркулиращата концентрация на ?OHB и намалява гликемията, но не оказва влияние нито върху обема на тумора, нито върху продължителността на преживяемостта при плъхове, носещи глиома (De Feyter et al., 2016). Предложен е глюкозен кетонов индекс като клиничен индикатор, който подобрява метаболитното управление на терапията на мозъчен рак, предизвикана от кетогенна диета при хора и мишки (Meidenbauer et al., 2015). Взети заедно, ролите на метаболизма на кетонните тела и кетонните тела в биологията на рака са примамливи, защото всеки от тях представлява поносими терапевтични възможности, но основните аспекти остават да бъдат изяснени, като ясни влияния възникват от матрица от променливи, включително (i) разлики между екзогенния кетон тела срещу кетогенна диета, (ii) тип ракови клетки, геномни полиморфизми, степен и стадий; и (iii) времето и продължителността на излагане на кетотично състояние.

Кетогенезата се създава от кетонни тела чрез разграждане на мастни киселини и кетогенни аминокиселини. Този биохимичен процес осигурява енергия на различни органи, по-специално мозъка, при условия на гладуване като отговор на липсата на кръвна глюкоза. Кетонните тела се произвеждат главно в митохондриите на чернодробните клетки. Докато други клетки са способни да извършват кетогенеза, те не са толкова ефективни в това, колкото чернодробните клетки. Тъй като кетогенезата се осъществява в митохондриите, нейните процеси се регулират независимо. Д-р Алекс Хименес DC, CCST Insight
Терапевтично приложение на кетогенна диета и екзогенни кетонни тела
Приложенията на кетогенните диети и кетонните тела като терапевтични инструменти също са възникнали в неракови контексти, включително затлъстяване и NAFLD/NASH (Browning et al., 2011; Foster et al., 2010; Schugar and Crawford, 2012); сърдечна недостатъчност (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); неврологични и невродегенеративни заболявания (Martin et al., 2016; McNally and Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang and Cheng, 2010; Yao et al., 2011); вродени нарушения на метаболизма (Scholl-B rgi et al, 2015); и изпълнение на упражненията (Cox et al., 2016). Ефикасността на кетогенните диети е особено оценена при терапията на епилептичен припадък, особено при резистентни към лекарства пациенти. Повечето проучвания оценяват кетогенните диети при педиатрични пациенти и разкриват до ~50% намаление на честотата на припадъци след 3 месеца, с подобрена ефективност при избрани синдроми (Wu et al., 2016b). Опитът е по-ограничен при епилепсия при възрастни, но подобно намаление е очевидно, с по-добър отговор при пациенти с симптоматична генерализирана епилепсия (Nei et al., 2014). Основните антиконвулсивни механизми остават неясни, въпреки че постулираните хипотези включват намалено използване на глюкоза/гликолиза, препрограмиран транспорт на глутамат, индиректно въздействие върху ATP-чувствителния калиев канал или аденозин A1 рецептор, промяна в експресията на изоформата на натриевия канал или ефекти върху циркулиращите хормони (включително лептин). Lambrechts et al., 2016; Lin et al., 2017; Lutas and Yellen, 2013). Остава неясно дали антиконвулсивният ефект се дължи основно на кетонните тела или на каскадните метаболитни последици от диетите с ниско съдържание на въглехидрати. Независимо от това, кетонните естери (вижте по-долу) изглежда повишават прага на гърчове при животински модели на провокирани припадъци (Ciarlone et al., 2016; D'Agostino et al., 2013; Viggiano et al., 2015).
Диети в стил Аткинс и кетогенни, ниско въглехидратни диети често се считат за неприятни и могат да причинят запек, хиперурикемия, хипокалцемия, хипомагнезиемия, да доведат до нефролитиаза, кетоацидоза, да причинят хипергликемия и да повишат концентрацията на циркулиращия холестерол и свободни мастни киселини, ; Kossoff and Hartman, 2001; Kwiterovich et al., 2012; Suzuki et al., 2003). Поради тези причини дългосрочното придържане представлява предизвикателства. Изследванията при гризачи обикновено използват отличително разпределение на макронутриентите (2002% kcal мазнини, 94% kcal въглехидрати, 1% kcal протеин, Bio-Serv F5), което провокира силна кетоза. Въпреки това, увеличаването на съдържанието на протеин, дори до 3666% kcal, значително намалява кетозата, а ограничаването на протеина от 10% kcal придава объркващи метаболитни и физиологични ефекти. Тази диетична формулировка също е изчерпана с холин, друга променлива, която влияе върху чувствителността към чернодробно увреждане и дори кетогенезата (Garbow et al., 5; Jornayvaz et al., 2011; Kennedy et al., 2010; Pissios et al., 2007; Schugar и др., 2013). Ефектите от дългосрочната консумация на кетогенни диети при мишки остават непълно дефинирани, но последните проучвания при мишки разкриват нормална преживяемост и липса на маркери за увреждане на черния дроб при мишки на кетогенни диети през целия им живот, въпреки че метаболизмът на аминокиселините, разходът на енергия и инсулиновата сигнализация бяха значително препрограмирани (Douris et al., 2013).
Механизмите, повишаващи кетозата чрез механизми, алтернативни на кетогенните диети, включват използването на поглъщани прекурсори на кетонни тела. Приложението на екзогенни кетонни тела може да създаде уникално физиологично състояние, което не се среща в нормалната физиология, тъй като концентрациите на циркулираща глюкоза и инсулин са относително нормални, докато клетките могат да спестят усвояването и използването на глюкоза. Самите кетонни тела имат кратък полуживот и поглъщането или вливането на натриева ?OHB сол за постигане на терапевтична кетоза провокира неблагоприятно натоварване с натрий. R/S-1,3-бутандиолът е нетоксичен диалкохол, който лесно се окислява в черния дроб, за да се получи d/l-?OHB (Desrochers et al., 1992). В различни експериментални контексти тази доза е прилагана ежедневно на мишки или плъхове в продължение на цели седем седмици, което води до циркулиращи концентрации на ?OHB до 5 mM в рамките на 2 часа след приложението, което е стабилно за поне допълнителни 3 часа (D' Агостино и др., 2013). Частично потискане на приема на храна е наблюдавано при гризачи, получаващи R/S-1,3-бутандиол (Carpenter and Grossman, 1983). В допълнение, три химически различни кетонни естера (KEs), (i) моноестер на R-1,3-бутандиол и d-pOHB (R-3-хидроксибутил R-pOHB); (ii) глицерил-трис-аОНВ; и (iii) R,S-1,3-бутандиол ацетоацетат диестер също са подробно изследвани (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al. ., 1995b; Kashiwaya et al., 2010). Присъщо предимство на първото е, че 2 мола физиологичен d-?OHB се произвеждат на мол KE, след хидролиза на естераза в червата или черния дроб. Безопасността, фармакокинетиката и толерантността са най-обстойно проучени при хора, поглъщащи R-3-хидроксибутил R-?OHB, в дози до 714 mg/kg, което води до циркулиращи концентрации на d-?OHB до 6 mM (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). При гризачи този KE намалява приема на калории и общия холестерол в плазмата, стимулира кафявата мастна тъкан и подобрява инсулиновата резистентност (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Последните открития показват, че по време на тренировка при тренирани спортисти, поглъщането на R-3-хидроксибутил R-?OHB намалява гликолизата на скелетните мускули и плазмените концентрации на лактат, повишава интрамускулното окисление на триацилглицерол и запазва съдържанието на мускулен гликоген, дори при едновременно поглъщане на инсулинова секреция (стимулирана секреция на инсулин Кокс и др., 2016). Необходимо е по-нататъшно развитие на тези интригуващи резултати, тъй като подобрението в изпълнението на упражненията за издръжливост се дължи предимно на силен отговор на KE при 2/8 субекта. Независимо от това, тези резултати подкрепят класическите проучвания, които показват предпочитание към окисление на кетон пред други субстрати (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley et al., 2003; Valente-Silva et al., 2015), включително по време на тренировка и че тренираните спортисти могат да бъдат по-подготвени да използват кетони (Johnson et al., 1969a; Johnson and Walton, 1972; Winder et al., 1974; Winder et al., 1975). И накрая, механизмите, които биха могли да подкрепят подобрената производителност на упражненията след равен прием на калории (диференциално разпределени между макронутриентите) и равни нива на консумация на кислород остават да бъдат определени.
Бъдеща перспектива
Веднъж до голяма степен стигматизиран като преливащ път, способен да натрупва токсични емисии от изгарянето на мазнини в състояния с ограничени въглехидрати (парадигмата на „кетотоксичната“), последните наблюдения подкрепят схващането, че метаболизмът на кетоновите тела изпълнява благотворна роля дори в състояния, натоварени с въглехидрати, отваряйки „кетохорметик“. хипотеза. Докато лесните хранителни и фармакологични подходи за манипулиране на метаболизма на кетоните го правят привлекателна терапевтична цел, агресивно поставени, но разумни експерименти остават както в основните, така и в транслационните изследователски лаборатории. Появиха се неудовлетворени нужди в областите на определяне на ролята на усвояването на кетонния метаболизъм при сърдечна недостатъчност, затлъстяване, NAFLD/NASH, диабет тип 2 и рак. Обхватът и въздействието на „неканоничните“ сигнални роли на кетонните тела, включително регулирането на PTMs, които вероятно се връщат назад и напред в метаболитни и сигнални пътища, изискват по-задълбочено изследване. И накрая, екстрахепаталната кетогенеза може да отвори интригуващи паракринни и автокринни сигнални механизми и възможности за повлияване на съвместния метаболизъм в нервната система и туморите за постигане на терапевтични цели.
Благодарности
Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/
Бележки под линия
В заключение, кетонните тела се създават от черния дроб, за да се използват като източник на енергия, когато в човешкото тяло няма достатъчно глюкоза. Кетогенезата възниква, когато има ниски нива на глюкоза в кръвта, особено след изчерпване на други клетъчни запаси от въглехидрати. Целта на статията по-горе беше да се обсъдят многоизмерните роли на кетонните тела в горивния метаболизъм, сигнализирането и терапията. Обхватът на нашата информация е ограничен до хиропрактика и проблеми със здравето на гръбначния стълб. За да обсъдите темата, моля, не се колебайте да попитате д-р Хименес или да се свържете с нас на 915-850-0900 .
Подготвен от д-р Алекс Хименес
Цитирано от: Ncbi.nlm.nih.gov/pmc/articles/PMC5313038/

Допълнителна дискусия на тема: „Остра болка в гърба
Заболявания на опорно-двигателния апарат е една от най-разпространените причини за инвалидност и пропуснати работни дни в световен мащаб. Болката в гърба се приписва на втората най-честа причина за посещения на лекар, превъзхождаща само инфекциите на горните дихателни пътища. Приблизително 80 процента от населението ще изпита болки в гърба поне веднъж през живота си. Гръбначният стълб е сложна структура, съставена от кости, стави, връзки и мускули, наред с други меки тъкани. Наранявания и/или влошени състояния, като напрхерния дискове, в крайна сметка може да доведе до симптоми на болки в гърба. Спортните наранявания или нараняванията при автомобилни катастрофи често са най-честата причина за болки в гърба, но понякога най-простите движения могат да имат болезнени резултати. За щастие, алтернативните възможности за лечение, като хиропрактика, могат да помогнат за облекчаване на болката в гърба чрез използване на корекции на гръбначния стълб и ръчни манипулации, като в крайна сметка подобряват облекчаването на болката.

EXTRA EXTRA | ВАЖНА ТЕМА: Препоръчителна El Paso, TX Chiropractor
***